
More Information
Submitted: December 15, 2020 | Approved: January 19, 2021 | Published: January 20, 2021
How to cite this article: Bajaj A. The coral crunch: Amyloidoma. Ann Dermatol Res. 2021; 5: 001-005.
DOI: 10.29328/journal.adr.1001013
Copyright License: © 2021 Bajaj A. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Antisecretory function; Cross-reactivity; Delayed reaction; Famotidine; Hypersensitivity; Maculopapular exanthema; Patch test; Ranitidine

The coral crunch: Amyloidoma
Anubha Bajaj*
Histopathologist, A.B. Diagnostics, New Delhi, India
*Address for Correspondence: Anubha Bajaj, Histopathologist, A.B. Diagnostics, New Delhi, India, Tel: 00911141446785; Email: [email protected]
Amyloidoma is an exceptional, progressive disorder demonstrating a characteristic accumulation of significant quantities of amyloid within soft tissues. Amyloidoma is additionally nomenclated as tumoural amyloidosis, nodular amyloid or localized amyloidosis. Furthermore, insulin-derived amyloidoma is referred to as insulin ball. Amyloid is a protein polymer configured of identical monomeric protein units wherein pathological variety is articulated from misfolded proteins. In excess > of twenty three subtypes of proteins can configure amyloid fibres in vivo. Extra-cellular or intra-cellular deposition of amyloid can modify normal organ function [1].
Amyloidosis is categorized into systemic and localized subtypes. Localized amyloidosis displays a localized mass effect and demonstrates a superior prognosis. Insulin-derived amyloidosis was initially documented by Storkel, et al. in 1983 who recognized accumulated insulin- amyloid fibrils in diabetic individuals subjected to continuous infusion of porcine insulin over a period of 5 weeks or more [1,2]. Amyloid nodules may be associated with systemic amyloidosis.
Disease pathogenesis
Amyloidosis is classified pertaining to cogent clinical and pathological criterion as
a) Primary amyloidosis demonstrating an absence of an identifiable cause
b) Secondary amyloidosis emerging as a consequence of chronic diseases such as tuberculosis or rheumatoid arthritis
c) Amyloidosis associated with multiple myeloma
d) Localized amyloidosis which characteristically displays absence of evident systemic incrimination or underlying chronic disease[1,3].Contemporary, chemical classification of amyloidosis is contingent to accumulated protein subtype and is characterized as amyloid associated (AA) or amyloid light- chain (AL) amyloidosis [1,3].
Systemic amyloidosis is denominated by diverse categories of amyloid accumulation
a) Amyloid associated (AA) amyloidosis is frequently discerned and exhibits precursor serum amyloid An incriminating the spleen, hepatic and renal parenchyma. Diverse rheumatologic disorders (rheumatoid arthritis, ankylosing spondylitis, juvenile idiopathic arthritis), autoimmune disorders (psoriasis, psoriatic arthritis) or haematological malignancies (Hodgkin’s and non Hodgkin’s lymphoma, multiple myeloma) are associated with systemic amyloidosis [3,4].
Previously treated mycobacterial infection or pulmonary tuberculosis and chronic inflammation arising due to repetitive respiratory tract infections may incur AA amyloidosis [3].
b) Light –chain (AL) amyloidosis is a variant of systemic amyloidosis where clonal immunoglobulin light- chains or light- chain fragments configure the precursor protein. AL amyloidosis ensues with production of aberrant antibodies within the bone marrow which are resistant to denaturation. Aforesaid antibodies configure amyloid and are deposited within soft tissues thereby engendering organ dysfunction. AL amyloidosis commonly implicates cardiac tissue, renal parenchyma, gastrointestinal tract, respiratory tract or peripheral nervous system.
c) Heavy chain (AH) amyloidosis is engendered by accumulation of fibrils of immunoglobulin heavy chain sequences [3,4]. Amyloidosis can also occur with deposition of transthyretin or beta-2 microglobulin thereby articulating transthyretin amyloidosis (ATTR) or beta-2 microglobulin amyloidosis (Abeta2M) or associated varieties of systemic amyloidosis [3,4]. Localized amyloidosis is configured by on site production and accumulation of amyloid substances in diverse tissues and organs. Mature plasma cells secrete abundant quantities of misfolded light chain immunoglobulins which accumulate at diverse sites [3,4].
Localized amyloidosis or “amyloidoma” represents a true neoplasm rather than a pseudo tumour and may be incurred by plasma cells. Specifically, clonal expansion of singular plasma cell is denominated with localized (AL) amyloidosis. Localized amyloidosis can appear within the urinary bladder, trachea, bronchi, larynx, gastrointestinal tract, orbit, tonsils, lymph nodes and cutaneous surfaces. Localized amyloidosis of the brain tissue typically engenders Alzheimer’s disease. Localized amyloidosis is frequently associated with Sjogren’s syndrome [3,4].
Precise aetiology of localized amyloidosis is obscure. As amyloidosis is usually articulated within mucous membranes, prolonged activation of tissue plasma cells due to exposure to environmental antigens is a preliminary step in disease generation. Reactive plasma cells exhibit enhanced production of amyloidogenic immunoglobulin light chains. Transformation of aforesaid immunoglobulin light chains into insoluble fibrils due to activation of tissue macrophages is a key mechanism in articulating the amyloidoma, a hypothesis which is validated by presence of several multinucleated giant cells within an amyloidoma [3,4]. Localized amyloidosis frequently involves the larynx and rarely implicates the tongue. Nodules may depict accumulated amyloid protein [3,4].
Disease characteristics
The asymptomatic, nodular tumefaction is usually discerned in adults and elderly population. Commonly incriminated sites are lower extremities, cutaneous surfaces or subcutaneous tissue. The condition is idiopathic or may arise as a consequence of multiple myeloma, plasmacytoid lymphoma and in individuals with chronic inflammation or on long-term dialysis [5,6].
Contemporary recommendations pertaining to categorization of amyloidosis are:
a) Congo red staining is contemplated as the gold standard for detection of amyloid.
b) Appropriate discernment of amyloid subtype requires adequate microscopic evaluation and immunohistochemistry. Singular, clinical assessment or deoxy ribonucleic acid (DNA) assay may be insufficient.
c) Competent immuno-histochemical analysis and confirmation with sophisticated techniques is necessitated [5,6].
Insulin- derived amyloidosis is commonly discerned in subjects with inadequate glycaemic control demonstrating glycosylated haemoglobin (HbA1c) levels between 7.6% to 15.5% and duration of insulin therapy varying from 4 years to 60 years. Insulin-derived amyloidosis is a disorder of enhancing prevalence and lesions are commonly discerned within the abdomen (70%), thigh (15%), upper extremity (13%), breast (1%) or inguinal lymph nodes (1%). Insulin-derived amyloidoma is discerned in 3.5% individuals subjected to aspiration of abdominal adipose tissue. Following pertinent insulin therapy, around 34% of insulin is absorbed when injected into the site of amyloidoma with consequent hyperglycaemia [3,4].
Mechanism of insulin metamorphosis into amyloid fibrils is inadequately posited and multiple pathways exist.
Disassociation of insulin hexamers and dimers into monomeric forms is a predominant step as configuration of fibrils is rapid with a dominant monomer. Also, occurrence of covalent dimers prohibits articulation of fibrils. Additionally, A and B chains of insulin molecule are amyloidogenic. Thus, localized, iatrogenic, insulin-induced amyloidoma is termed as AIns amyloidosis [3,4].
Primary cutaneous amyloidoma or drug induced amyloidosis can be induced by Enfuviritide, an anti- retroviral agent employed for treating individuals with human immune deficiency virus (HIV) infection. The drug induces accumulation of amyloid (AEnf amyloid) at injection sites, akin to insulin or subcutaneously administered protein and peptide agents [3,5].
Clinical elucidation
Primary cutaneous amyloidosis principally delineates categories such as
• lichen (papular) amyloidosis with altered keratin
• macular amyloidosis with altered keratin
• nodular amyloidosis with light chains (AL amyloid).
Localized amyloidosis appears within the head and neck region and is predominantly discerned in larynx or trachea whereas amyloidoma or diffuse enlargement of the tongue is exceptional. A polypoid tumefaction of variable magnitude protruding into superimposed cutaneous or mucosal surface can be exhibited [6,7].
Insulin- derived amyloidosis exhibits a solitary (90%), firm or soft subcutaneous nodule of variable magnitude appearing at the site of insulin injections [6,7].
Cogent clinical history, physical features and magnetic resonance imaging (MRI) of incriminated sites along with histological assessment in combination with Congo red stain can appropriately discern localized light- chain (AL) or amyloid associated (AA) amyloidosis [6,7].
Histological elucidation
On gross examination, a soft, yellowish, non friable neoplasm of magnitude varying from one centimetre to three centimetres is delineated.
Soft tissue fragments are translucent and display a waxy, firm, starch- like consistency [7,8].
Microscopy depicts an extra-cellular or sub-epithelial deposit of an acellular, eosinophilic, homogenous or matrix-like material. Accompanying inflammatory infiltrate is sparse and composed of lymphocytes and mature plasma cells [7,8].
Insulin-derived amyloidosis demonstrates accumulation of a dense, eosinophilic, amorphous substance within the subcutaneous tissue.
Morphologically, well circumscribed aggregates of amyloid are frequently intermingled with small lymphocytes (T lymphocytes) and plasma cells. Amyloidoma can frequently evoke a granulomatous reaction composed of epithelioid cells admixed with disseminated multinucleated giant cells. Additionally, foci of calcification and ossification are discerned [7,8].
Tumefaction exemplifies islands of amorphous, eosinophilic material admixed with mature plasma cells and multinucleated giant cells. Foci of calcification and metaplastic bone are disseminated within the cellular component. Walls of enmeshed vascular articulations are imbued with amyloid substance [7,8].
Amyloid tissue can be appropriately discerned with periodic acid Schiff’s stain with pre-digestion with diastase (PASD+) [7,8]. Congo red stain delineates sub-epithelial deposits of reddish, homogenous substance which demonstrates an apple- green birefringence under polarized light. Birefringence persists following pre-treatment with potassium permanganate in primary, light chain amyloidosis (AL) [7,8].
Ultrastructural examination depicts accumulated amyloid fibrils [6] (Figures 1-8).
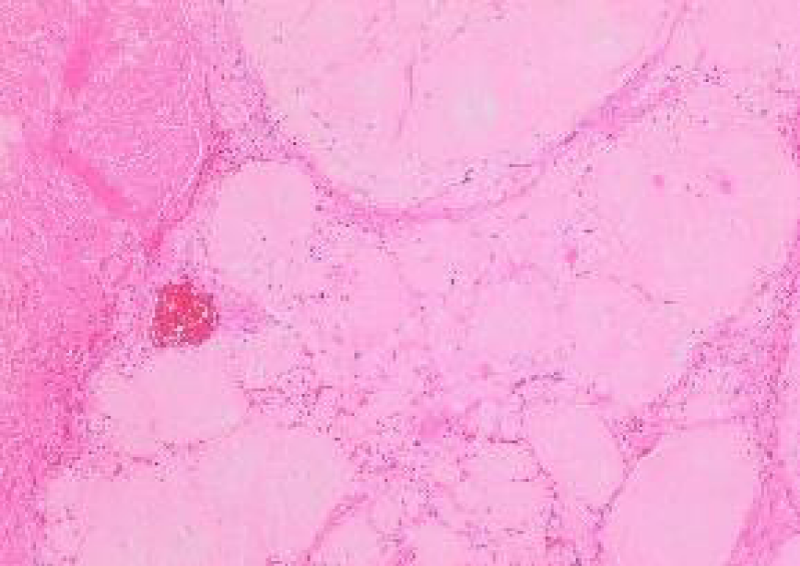
Figure 1: Amyloidoma of trigeminal nerve displaying nodules of acellular, amorphous, eosinophilic material with circumscribing collagen. Courtesy: Cureus.com
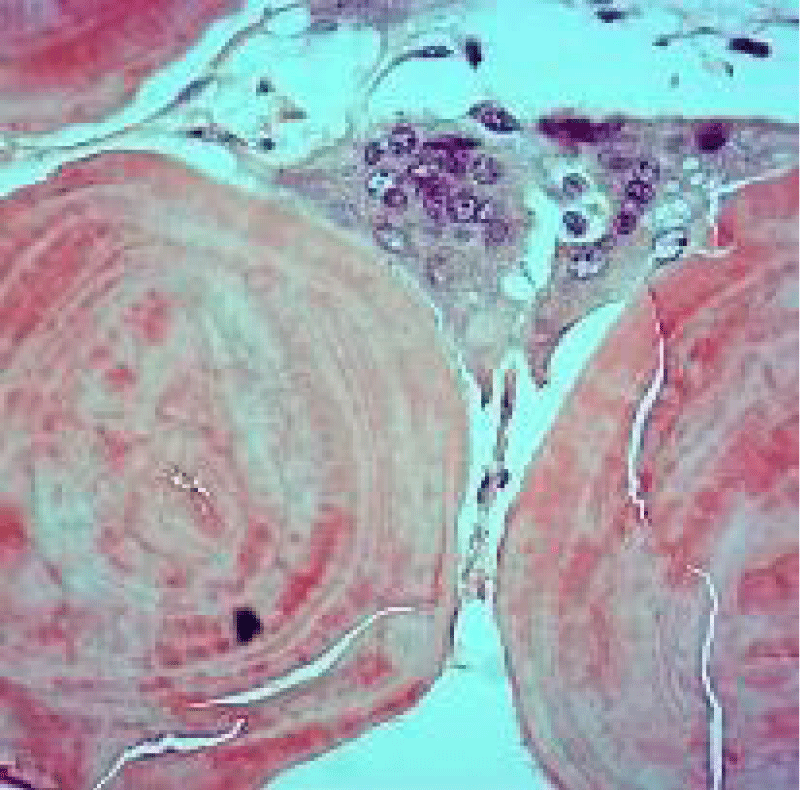
Figure 2: Amyloidoma of pulmonary parenchyma depicting globules of abundant, amorphous, eosinophilic amyloid interspersed with multinucleated giant cells. Courtesy: Pathology outlines
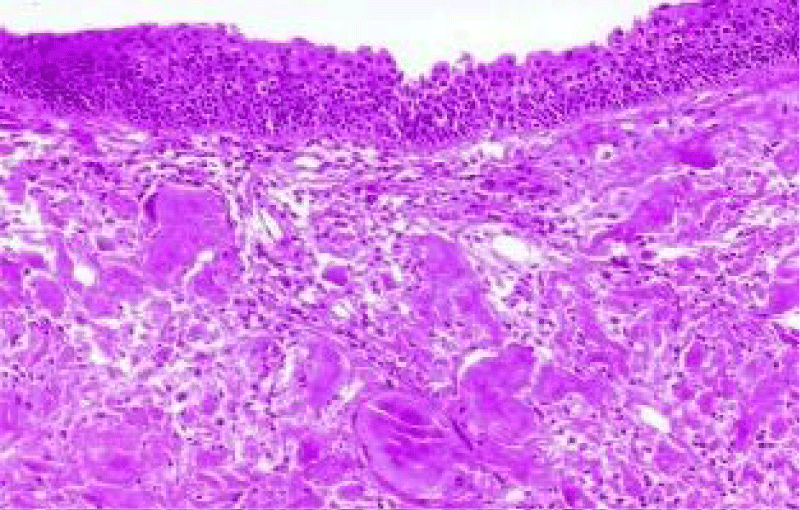
Figure 3: Amyloidoma of larynx exemplifying aggregates of amorphous, eosinophilic substance superimposed by pseudostratified columnar epithelium. Courtesy: Nature.com
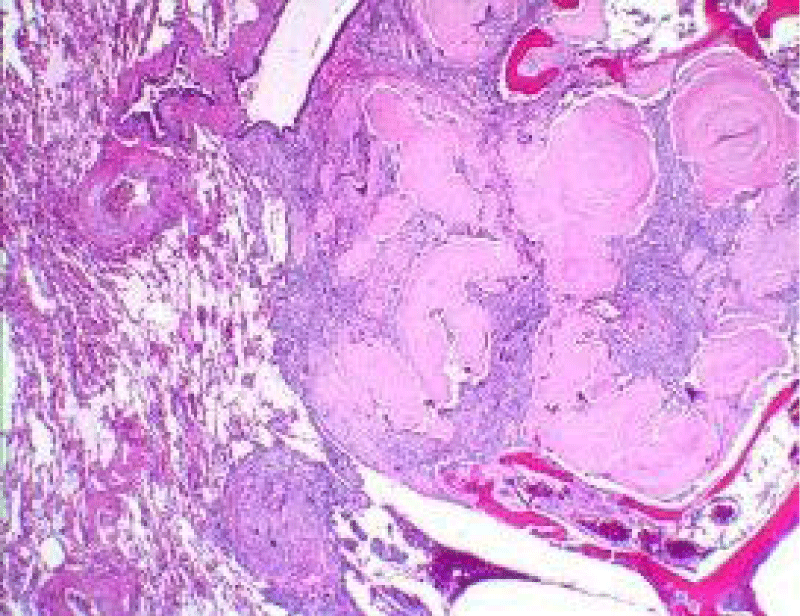
Figure 4: Amyloidoma of soft tissue delineating accumulation of eosinophilic, acellular amyloid material intermixed with inflammatory exudate of plasma cells and lymphocytes. Courtesy: Pathology apps
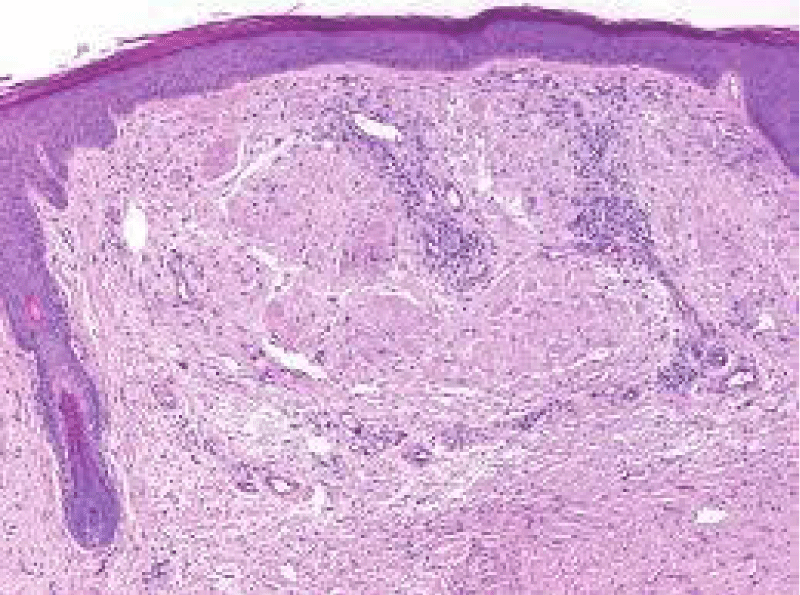
Figure 5: Amyloidoma of cutaneous surface with nodular deposit of acellular, amorphous, eosinophilic substance intermixed with mature lymphocytes and collagen fibres. Courtesy: Wiley online library
Figure 6: Amyloidoma of the thoracic activity enunciating acellular deposits of eosinophilic material, multinucleated giant cells, lymphocytes and mature plasma cells. Courtesy: Journal of Neurology
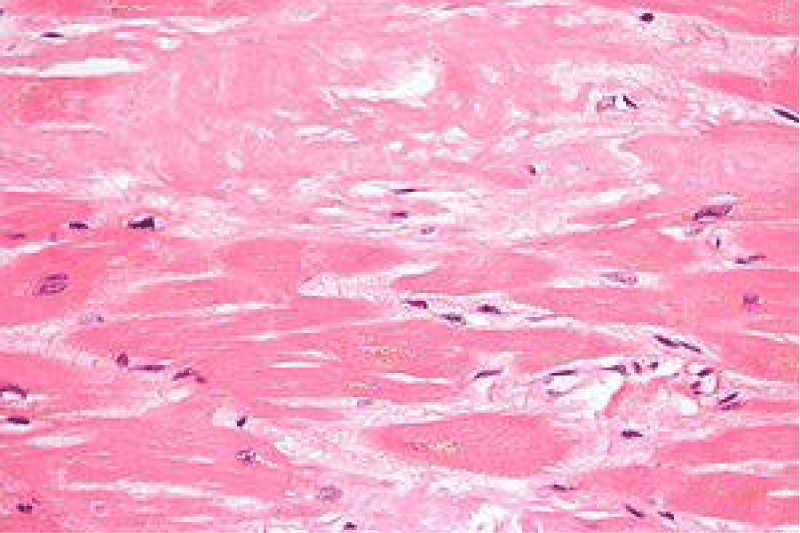
Figure 7: Amyloidoma of the cardiac muscle delineating localized accumulation of acellular, eosinophilic amyloid substance and disseminated lymphocytes. Courtesy: Research Gate
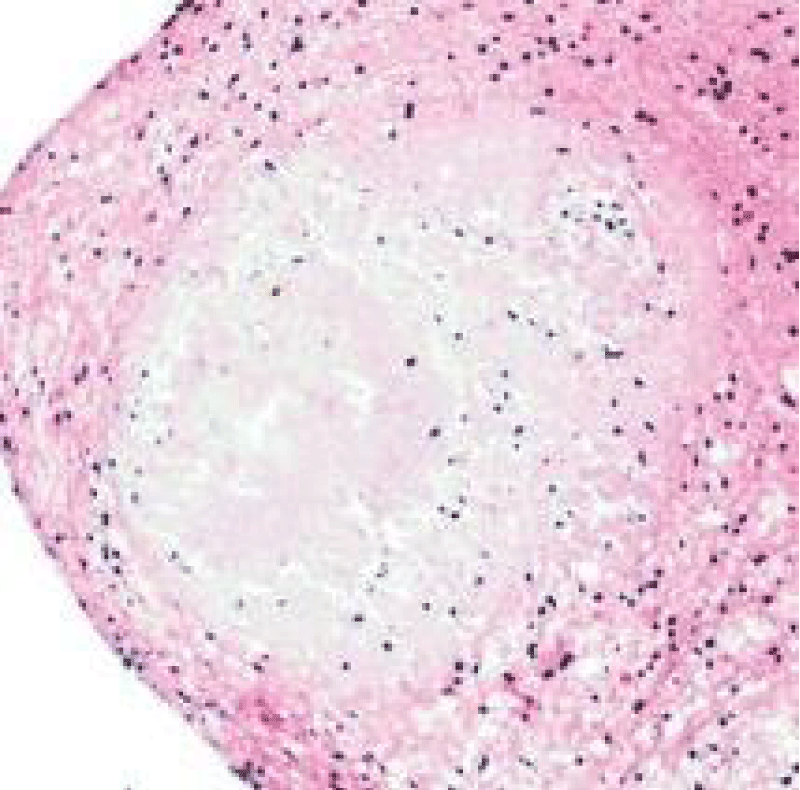
Figure 8: Amyloidoma of the brain depicting localized accumulation of acellular, eosinophilic amyloid material interspersed with chronic inflammatory cells as lymphocytes and plasma cells. Courtesy: Libre pathology
Immune histochemical elucidation
Amyloid stained with Congo red demonstrates a glassy, salmon or pink, amorphous, congophilic material which depicts an apple- green birefringence under polarized microscopy. The neoplasm is immune reactive to CD20, thereby indicating a B lymphocyte origin [3,4].
In insulin- derived amyloidosis, accumulated amyloid is immune reactive to anti-insulin antibodies. The nodule is immune reactive to insulin and demonstrates darkly stained, intense deposits of insulin which are indicative of insulin derived amyloid [3,4]. Plasma cells depict a clonal restriction with exemplification of either kappa or lambda light chains. Immunohistochemistry with kappa or lambda light chains may be adopted in order to confirm a clonal disorder as light chain amyloidosis (AL). Deposition of amyloid a protein confirms occurrence of amyloid associated (AA) amyloidosis. However, aforesaid evaluation is imprecise and nonspecific [7,8].
Differential diagnosis
Amyloidoma mandates a segregation from neoplasms such as
• Primary pulmonary lymphoma with amyloid production depicts an estimated beneath < 1% of pulmonary lymphomas with deposition of amyloid. Generally, individuals exceeding 70 years are incriminated. Subjects with marginal zone or small lymphocytic lymphoma/ chronic lymphatic leukaemia (SLL/ CLL) frequently denominate lymphatic tracking or reactive lymphoid follicles along with evenly disseminated, mature, small lymphocytes within the nodule in combination with pleural infiltration, features which are contemplated as specific for pulmonary lymphoma [9,10].
• Hyalinising granuloma is accompanied by history of exposure to fungal agents such as Histoplasma or Mycobacterium tuberculosis. Collagen contained within the lesions may not stain with Congo red [9,10].
Insulin-derived amyloidoma appearing at the site of insulin injection requires a segregation from
• Lipo-hypertrophy which is a condition which clinically simulates insulin- induced amyloidosis. Clinical distinction may not be possible although insulin lesions are firm in consistency. Individuals with lipo-hypertrophy are associated with declining insulin efficacy. In contrast to insulin-induced amyloidosis, magnitude of lesions of focal insulin-associated lipo-hypertrophy decimate with discontinuation of insulin injections [9,10].
Investigative assay
Implicated individuals can be subjected to plain radiography of the chest, echocardiography, hepatic fibro-scan, assessment of N Terminal - pro-brain natriuretic peptide, routine haematological and biochemical parameters, immunoelectrophoresis and segregation from adjunctive, simulating conditions [11,12].
Tumour nodule can be discerned upon plain radiograph [11].
Clinical suspicion of amyloidosis mandates adequate histological confirmation. Cogent tissue specimens can be obtained from sites incriminated with localized amyloidosis and competent microscopy is sufficient to establish the disease. Upon staining with haematoxylin and eosin, amyloid appears as a homogenous, eosinophilic, amorphous substance. Subsequent staining with Congo red and examination with polarized light reveals an apple- green birefringence [11,12].
With confirmation of amyloidosis, categorization of subtype is required which is achieved by immunoelectrophoresis of serum or urine. Aforesaid procedure is also adopted to detect a clonal disorder [11,12].
Ultrasonography (US) can appropriately define breast amyloidoma and is beneficial in obtaining guided tissue samples. Ultrasound can suitably denominate extent of soft tissue incrimination and recommended, pertinent surgical excision [11,12].
Magnetic resonance imaging (MRI) of incriminated sites such as head and neck may demonstrate the nodular lesion. Tumour infiltration into adjacent soft tissue is absent [11,12].
Computerized tomography (CT) and magnetic resonance imaging (MRI) may assist demarcation between insulin- derived amyloidosis and lipo-hypertrophy. Insulin-induced amyloidoma can be adequately discerned with tissue sampling or aspiration followed by surgical resection of the nodule [11,12].
Therapeutic options
Prognostic outcome of light chain (AL) amyloidosis is contingent to extent of disease. Localized amyloidosis demonstrates an excellent prognosis and progression to systemic amyloidosis is infrequent [11,12].
On account of superior prognosis, typically the neoplasm may not progress to a lymphoproliferative disorder. Therefore, localized amyloidosis may not mandate systemic therapy [11,12].
Therapeutic management can be supportive or localized. Common treatment strategies are localized surgical extirpation, ablation or clinical observation of disease. Surgical extermination or ablation of amyloidoma is a preferred treatment modality and is appropriately adopted for management of associated clinical manifestations such as patency of airways [11,12].
Surgical extraction of insulin- derived amyloidosis may be accompanied by extrusion of acellular amyloid from the nodule. Although surgical resection of the neoplasm is an optimal treatment strategy, adequate clinical monitoring can also be efficaciously adopted. Clinical follow up for a duration of two years is usually adequate [11,12].
Tumefaction can be eradicated by laser or electrocautery. Adoption of suitable technique of amyloid extrusion can induce significant de-bulking and resolution of amyloidoma [11,12].
Uniform rotation of sites of insulin injection can be advantageously utilized to circumvent development of amyloidoma. Tumour reoccurrence is frequent [11,12].
- Musat G, Evsei A, Calina D, Docea AO, Doukas SG, et al. Rare amyloidoma of the tongue base – a case report and review of the literature. Mol Clin Oncol. 2020; 12: 258-262. PubMed: https://pubmed.ncbi.nlm.nih.gov/32064103/
- Storkel S, Schneider HM, Müntefering H, Kashiwagi S. Iatrogenic, insulin-dependent, local amyloidosis. Lab Invest. 1983; 48; 108-111. PubMed: https://pubmed.ncbi.nlm.nih.gov/6337294/
- Gertz MA. Immunoglobulin light chain amyloidosis: 2020 update on diagnosis, prognosis, and treatment. Am J Hematol. 2020; 95: 848-860. PubMed: https://pubmed.ncbi.nlm.nih.gov/32267020/
- Samlaska C, Reber S. Murry T. Insulin-derived amyloidosis – the insulin ball, amyloidoma. JAAD Case Rep. 2020; 6; 351-353. PubMed: https://pubmed.ncbi.nlm.nih.gov/32258319/
- Geller HI, Singh A, Mirto TM, Padera R, Mitchell R, et al. Prevalence of monoclonal gammopathy in wild-type transthyretin amyloidosis. Mayo Clin Proc. 2017; 92: 1800-1805. PubMed: https://pubmed.ncbi.nlm.nih.gov/29202938/
- Asiri MMH, Engelsman S, Eijkelkamp N, Höppener JWM. Amyloid Proteins and Peripheral Neuropathy. Cells. 2020; 9: 1553. PubMed: https://pubmed.ncbi.nlm.nih.gov/32604774/
- Westermark P. Localized AL amyloidosis- a suicidal neoplasm? Ups J Med Sci. 2012; 117: 244-250. PubMed: https://pubmed.ncbi.nlm.nih.gov/22335280/
- O’Reilly A, D’Souza A, Lust J, Price D. Localized tongue amyloidosis – a single institutional case series. Otolaryngol Head Neck Surg. 2013: 149: 240-244. PubMed: https://pubmed.ncbi.nlm.nih.gov/23715681/
- Hazenberg BP. Amyloidosis-a clinical review. Rheum Dis Clin North Am. 2013; 39: 323-345. PubMed: https://pubmed.ncbi.nlm.nih.gov/23597967/
- Balwani MR, Kute VB, Shah PR, Wakhare P, Trivedi HL. Secondary renal amyloidosis in a patient of pulmonary tuberculosis and common variable deficiency. J Nephropharmacol. 2015; 4: 69-71. PubMed: https://pubmed.ncbi.nlm.nih.gov/28197481/
- Charlot M, Seldin DC, O'hara C, Skinner M, Sanchorawala V. Localized amyloidosis of the breast – a case series. Amyloid. 2011: 18; 72-75. PubMed: https://pubmed.ncbi.nlm.nih.gov/21501022/
- Kubota K, Ito R, Furudate K, Kon T, Nakagawa H, et al. Localized AL amyloidosis of the tongue-a case report and literature review. J Oral Maxillofac Surg Med Pathol. 2017; 29: 142-145